基于预训练语言模型提示的药物靶标相互作用预测方法
作者: 任鹏 段乐乐
摘要:药物靶标相互作用的准确预测对加速新药研发至关重要。近年来,基于序列的药物靶标相互作用预测方法受到广泛关注,并在预测性能上取得了显著进展。然而,直接采用Transformer架构的序列编码器通常面临参数量大、计算复杂度高的问题,从而限制了其实际应用。为解决这一问题,提出了一种基于提示的学习策略。该策略通过引入可学习的门控参数,将预训练语言模型的通用知识作为提示,动态适配到学生模型的特定任务知识,从而优化序列的特征表示。实验结果表明,该方法在减少模型参数的同时,提升了预测性能,能够实现高效且准确的药物靶标相互作用预测。
关键词:新药研发;药物靶标相互作用;深度学习;提示学习;预训练语言模型;教师学生架构
中图分类号:TP181 文献标识码:A
文章编号:1009-3044(2025)07-0034-04
开放科学(资源服务) 标识码(OSID)
0 引言
新药研发是一个复杂且耗时的过程,包括靶标识别、先导化合物发现、临床前研究和临床试验等多个阶段。然而,根据报告显示[1],大多数药物研发项目在临床试验阶段以失败告终,其主要原因在于靶标验证的不足。这一现象表明,针对特定靶标进行精确的药物筛选是新药研发迈向成功的关键第一步。
在此背景下,药物靶标相互作用(Drug-Target Interaction, DTI) 预测成为加速新药研发进程的重要研究方向。药物靶标相互作用预测的核心目标是识别能够与疾病靶标结合的药物分子,从而通过抑制或激活特定生物过程改善疾病症状。快速且准确地筛选出具有生物活性的药物分子,将显著提高新药研发的效率。
传统的湿实验方法尽管能够提供可靠的药物靶标相互作用结果,但这些方法依赖生物化学和生物物理实验技术,面临高成本、长周期等限制,难以在大规模数据中推广应用。相比之下,计算方法凭借其处理大规模数据的效率,可迅速缩小候选药物的搜索范围,并在实验验证之前筛选出潜在药物分子,因此受到广泛关注。尤其是近年来基于深度学习的计算方法,因其显著的性能提升而备受青睐[2]。此类方法通常将药物分子SMILES字符串和蛋白质氨基酸序列视为类似自然语言的序列,利用Transformer架构对其进行建模,自动学习药物分子和蛋白质靶标的特征表示。然而,这种方法存在一个局限性,即其计算复杂度随输入序列长度的增加呈二次方增长[3]。尤其是蛋白质氨基酸序列由于其固有的复杂性和较长的长度,使得有效建模需要大规模参数化的模型,进而导致推断时间显著增加。
为解决上述问题,本文提出了一种基于预训练语言模型提示的药物靶标相互作用预测方法。该方法的核心在于采用两个预训练语言模型作为教师模型,分别用于药物分子和蛋白质靶标的特征提取。通过提前缓存教师模型输出的特征表示,并将这些表示作为简化学生模型的提示,将教师模型的通用知识适配到与药物靶标相互作用预测任务相关的学生模型的特定知识。这种基于提示的知识适配方法显著减少了模型的参数规模,提高了计算效率,同时提升了药物靶标相互作用预测的准确性。
1 相关研究
1.1 分子语言模型
基于大规模分子数据集预训练的分子语言模型已成为研究分子结构与功能关系的重要工具。这类模型借鉴自然语言处理领域的自监督学习方法,以分子的SMILES字符串作为输入,能够从中自动学习深层次的分子特征。其中,ChemBERTa[4]和MoLFormer[5]模型具有代表性。ChemBERTa采用RoBERTa架构,通过捕捉SMILES序列中的上下文信息,提供了强大的分子表示能力,显著提高了分子性质预测的精度。相较于ChemBERTa,MoLFormer利用更大规模的数据集和更复杂的预训练策略,进一步增强了分子表示的适应性,可以更充分地捕捉分子的化学和结构信息。这些分子语言模型不仅在分子性质预测中表现出色,还为分子与蛋白质靶标的相互作用建模提供强大的支持,使得模型能够有效地从分子序列中提取高质量的语义特征表示,为药物靶标相互作用的精确预测奠定基础。
1.2 蛋白质语言模型
蛋白质语言模型为探索蛋白质结构与功能关系开辟了全新路径。这些模型能够从蛋白质序列中提取复杂模式与特征,极大地推动了生物信息学领域的研究进展。以ProtBERT[6]和ESM-2[7]为代表的两种模型在不同任务中展现出了卓越性能。ProtBERT基于BERT结构,利用双向编码机制获取蛋白质序列的丰富语义表示,在序列注释和功能预测等任务中表现出色。与ProtBERT相比,ESM-2通过扩展模型规模和训练数据量,并整合进化信息,大幅提升了对蛋白质序列的建模能力。其能够端到端地从初级序列直接生成原子级分辨率的三维结构,大幅简化了特征工程步骤,并显著提高了计算效率和自动化水平。在药物靶标相互作用预测任务中,使用蛋白质语言模型处理复杂的蛋白质序列,可提供更好的蛋白质特征表示,从而增强药物与靶标结合的预测能力。
2 方法
本研究将药物分子的SMILES序列与蛋白质的氨基酸序列作为输入,目标是在药物靶标相互作用任务中预测其交互概率[p∈[0,1]]。直接使用语言模型编码序列在计算效率和预测准确度方面表现有限。为此,本文提出了一种新的药物靶标相互作用预测模型,其架构如图1所示。
该模型主要由三大模块构成:药物编码器、蛋白质编码器和分类预测器。药物编码器从SMILES序列中提取特征,包括ChemBERTa教师模型和一个简化的学生模型;蛋白质编码器采用类似架构,从蛋白质序列中提取特征,由ESM-2教师模型和对应的学生模型构成;分类预测器则整合上述模块输出的特征,预测药物靶标对的相互作用概率值。
本方法通过预训练语言模型提供对序列的一般性理解,将教师模型输出视为通用知识,并以此作为提示,指导学生模型从序列中学习特定任务相关知识,提升泛化性能。
2.1 药物编码器
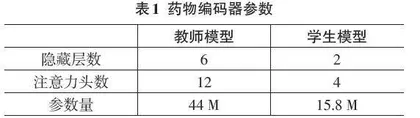
药物编码器对输入的SMILES序列进行编码,将其映射为特征向量。该模块包括教师模型与学生模型。教师模型选用ChemBERTa,基于PubChem数据集的SMILES序列进行预训练,能够捕获复杂的分子结构信息,并生成丰富的语义特征表示。该模型具有6个隐藏层和12个注意力头,输出特征维度为768。为提高特征表达的整体性,降低对序列长度的敏感度,本研究采用最后一个隐藏层所有词元向量的均值作为输出特征,而非特定类别词元(class token) 的向量。学生模型架构与ChemBERTa类似,仅减少了隐藏层数和注意力头数,其余超参数保持一致,使其参数量约为教师模型的36%。两者参数的详细对比见表1。
由于直接对大语言模型进行全参数微调在计算资源受限的情况下不现实,本文提出基于提示的训练策略:将教师模型的输出缓存在本地磁盘中,训练过程中冻结教师模型,仅更新学生模型参数。通过可学习的门控参数[α],提供一种动态知识适配机制。具体而言,在训练过程中,门控参数会根据梯度优化的结果自动调整其值,并以加权求和的形式将教师模型输出的通用知识与学生模型输出的特定知识相结合。药物编码过程形式化为:
[zd=(1-α)⋅gd[xd]+α⋅hd[xd]] (1)
式中:[xd]为输入的SMILES序列,其长度上限为512个词元,超过将被截断,短于则被填充。[gd[⋅]]和[hd[⋅]]分别代表药物编码器的教师模型和学生模型,用于编码给定的输入序列。参数[α]通过服从均匀分布[Uniformα[0,1]]随机初始化。在训练期间,若参数[α]值升高,表明编码器更倾向学生模型的特定知识。相反,若参数[α]值降低,表示编码器更多依赖教师模型的通用知识。
最终,药物特征表示[zd]经归一化层[Norm[⋅]]和线性变换函数[f[⋅]]进行维度对齐,从768维转换为512维:
[z′d=f[Norm[zd]]] (2)
2.2 蛋白质编码器
蛋白质编码器与药物编码器结构类似,由教师模型和学生模型组成,负责对蛋白质氨基酸序列进行特征提取。教师模型选用ESM-2,基于UniRef50数据集预训练,具有33个隐藏层和20个注意力头,参数量达650 M,能够生成高质量的蛋白质特征表示,输出特征维度为1 280。学生模型在架构设计上与教师模型保持一致,但显著降低了隐藏层数和注意力头数,参数量仅为教师模型的9.5%。两者具体参数对比见表2。
类似于药物编码器,教师模型在训练期间被冻结,仅提供提示信息。通过可学习的门控参数[β],融合教师模型的通用知识和学生模型的特定知识:
[zp=(1-β)⋅gp[xp]+β⋅hp[xp]] (3)
式中:[xp]表示输入的蛋白质序列,其长度固定为1 024个词元。[gp[⋅]]和[hp[⋅]]分别为蛋白质编码器的教师模型和学生模型,用于编码给定的蛋白质序列。参数[β]初始化方式和作用都与[α]相同。
蛋白质特征表示[zp]经归一化层[Norm[⋅]]和线性变换函数[f[⋅]]处理后,从1 280维转换为512维特征表示:
[z′p=f[Norm[zp]]] (4)
2.3 分类预测器
分类预测器模块接收维度为1 024的结合特征表示,该向量由对齐后的药物和蛋白质特征表示拼接得到。模块由三层顺序堆叠的全连接块组成,每层包括全连接层、GELU激活函数和丢弃层。全连接层的隐藏单元数依次为2 048、1 024和512,丢弃率均设为0.1。最终输出为预测的交互概率值。
3 实验
3.1 数据集
为了评估模型性能,本文选用先前研究[8]提供的两个基准数据集:BindingDB[9]和BioSNAP[10]。每个数据集按7∶1∶2的比例随机划分为训练集、验证集和测试集。BindingDB数据集可通过公开网络获取,主要包含药物靶标蛋白质与类药小分子之间相互作用的数据,其中包括经实验验证的结合亲和力值。BioSNAP数据集最初仅包含正类相互作用样本对,后在MolTrans[11]研究中,通过从未观测过的药物靶标对中采样补充了负类样本对,最终形成了正负样本数量相等的平衡数据集,这也是本文使用的数据集版本。两个基准数据集的统计信息详见表3。
3.2 评估指标
药物靶标相互作用预测任务属于二分类问题,本文主要采用两个评估指标:受试者工作特征曲线下面积(Area Under the Receiver Operating Characteristic Curve, AUROC) 和精确率-召回率曲线下面积(Area Under the Precision-Recall Curve, AUPRC) 。AUROC衡量了模型在不同决策阈值下区分正负样本的能力,值越大,说明模型区分能力越强。AUPRC适用于正负样本比例失衡的场景,直接反映模型对正类样本的检测性能,不受负类样本比例的影响。
此外,本文还引入了准确率(Accuracy) 、敏感性(Sensitivity) 和特异性(Specificity) 作为补充指标。这些指标均基于最佳F1分数的决策阈值计算,用于更全面地评估模型性能。
3.3 实验设置
实验在单块显存容量为24GB的NVIDIA A10显卡上进行,基于PyTorch深度学习框架实现,语言模型使用Transformers库构建。为提高数据处理的灵活性和训练流程的管理效率,实验还使用了PyTorch Lightning库组织训练、验证和测试阶段。
具体超参数配置如表4所示,两个数据集使用相同的设置,并未进行单独调参。优化器选用AdamW,同时采用余弦退火学习率调整策略,以促进模型的有效收敛并提升最终性能。